Info
cellxgene_
Eraslan et al. (2022)
2.99 GiB
02-02-2024
209126 × 28094
Quick links
Used in
No related benchmarks found.
cellxgene_
Eraslan et al. (2022)
2.99 GiB
02-02-2024
209126 × 28094
No related benchmarks found.
CREATED
02-02-2024
DIMENSIONS
209126 × 28094
Understanding the function of genes and their regulation in tissue homeostasis and disease requires knowing the cellular context in which genes are expressed in tissues across the body. Single cell genomics allows the generation of detailed cellular atlases in human tissues, but most efforts are focused on single tissue types. Here, we establish a framework for profiling multiple tissues across the human body at single-cell resolution using single nucleus RNA-Seq (snRNA-seq), and apply it to 8 diverse, archived, frozen tissue types (three donors per tissue). We apply four snRNA-seq methods to each of 25 samples from 16 donors, generating a cross-tissue atlas of 209,126 nuclei profiles, and benchmark them vs. scRNA-seq of comparable fresh tissues. We use a conditional variational autoencoder (cVAE) to integrate an atlas across tissues, donors, and laboratory methods. We highlight shared and tissue-specific features of tissue-resident immune cells, identifying tissue-restricted and non-restricted resident myeloid populations. These include a cross-tissue conserved dichotomy between LYVE1- and HLA class II-expressing macrophages, and the broad presence of LAM-like macrophages across healthy tissues that is also observed in disease. For rare, monogenic muscle diseases, we identify cell types that likely underlie the neuromuscular, metabolic, and immune components of these diseases, and biological processes involved in their pathology. For common complex diseases and traits analyzed by GWAS, we identify the cell types and gene modules that potentially underlie disease mechanisms. The experimental and analytical frameworks we describe will enable the generation of large-scale studies of how cellular and molecular processes vary across individuals and populations.
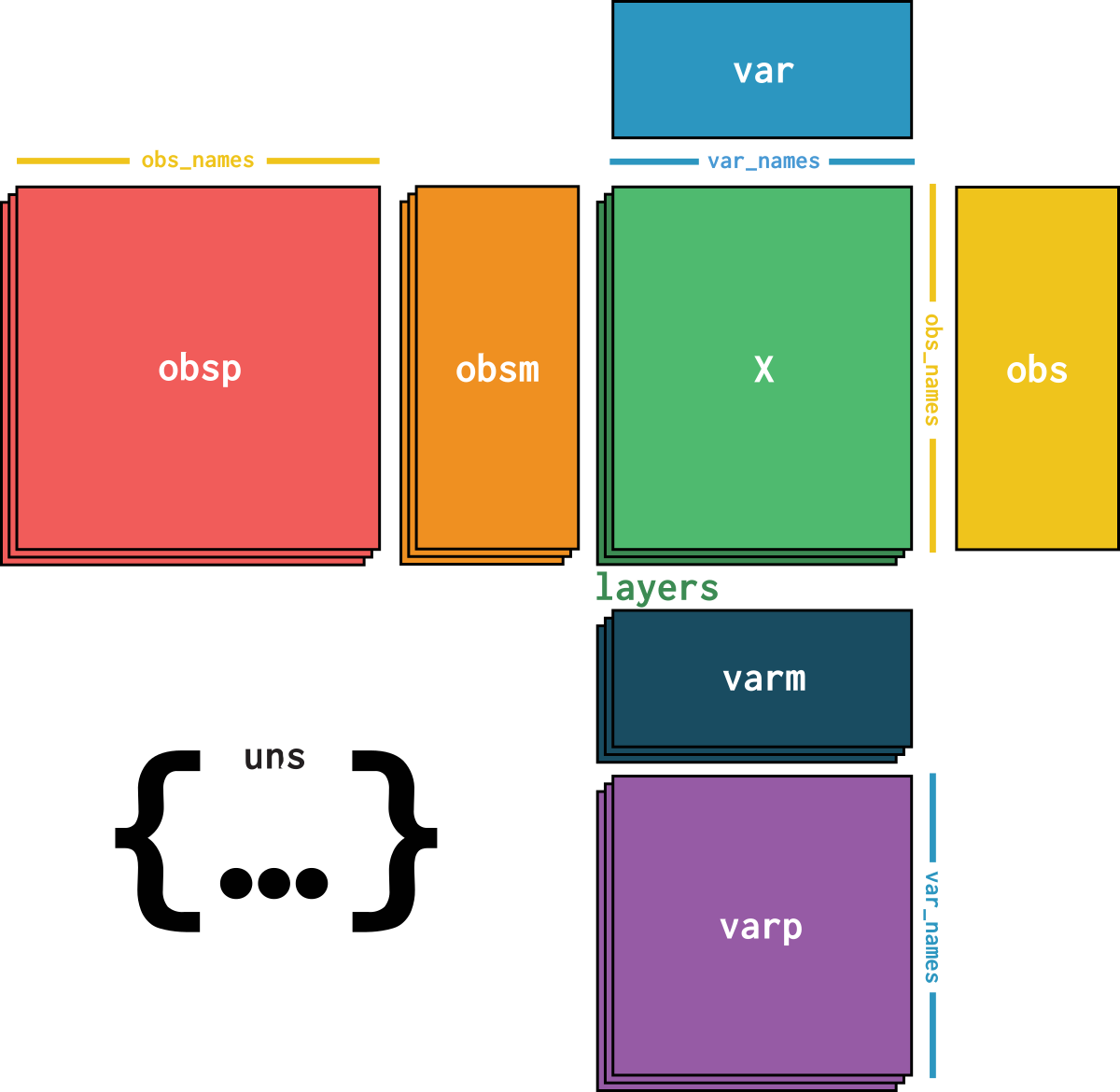
dataset is an AnnData object with n_obs × n_vars = 209126 × 28094 with slots:
soma_joinid, dataset_id, assay, assay_ontology_term_id, cell_type, cell_type_ontology_term_id, development_stage, development_stage_ontology_term_id, disease, disease_ontology_term_id, donor_id, is_primary_data, self_reported_ethnicity, self_reported_ethnicity_ontology_term_id, sex, sex_ontology_term_id, suspension_type, tissue, tissue_ontology_term_id, tissue_general, tissue_general_ontology_term_id, batch, size_factorssoma_joinid, feature_id, feature_name, hvg, hvg_scoreknn_connectivities, knn_distancesX_pcapca_loadingscounts, normalizeddataset_description, dataset_id, dataset_name, dataset_organism, dataset_reference, dataset_summary, dataset_url, knn, normalization_id, pca_variance| Name | Description | Type | Data type | Size |
|---|---|---|---|---|
| obs | ||||
assay
|
Type of assay used to generate the cell data, indicating the methodology or technique employed. |
vector
|
category
|
209126 |
assay_
|
Experimental Factor Ontology (EFO:) term identifier for the assay, providing a standardized reference to the assay type.
|
vector
|
category
|
209126 |
batch
|
A batch identifier. This label is very context-dependent and may be a combination of the tissue, assay, donor, etc. |
vector
|
category
|
209126 |
cell_
|
Classification of the cell type based on its characteristics and function within the tissue or organism. |
vector
|
category
|
209126 |
cell_
|
Cell Ontology (CL:) term identifier for the cell type, offering a standardized reference to the specific cell classification.
|
vector
|
category
|
209126 |
dataset_
|
Identifier for the dataset from which the cell data is derived, useful for tracking and referencing purposes. |
vector
|
category
|
209126 |
development_
|
Stage of development of the organism or tissue from which the cell is derived, indicating its maturity or developmental phase. |
vector
|
category
|
209126 |
development_
|
Ontology term identifier for the developmental stage, providing a standardized reference to the organism’s developmental phase. If the organism is human (organism_ontology_term_id == 'NCBITaxon:9606'), then the Human Developmental Stages (HsapDv:) ontology is used. If the organism is mouse (organism_ontology_term_id == 'NCBITaxon:10090'), then the Mouse Developmental Stages (MmusDv:) ontology is used. Otherwise, the Uberon (UBERON:) ontology is used.
|
vector
|
category
|
209126 |
disease
|
Information on any disease or pathological condition associated with the cell or donor. |
vector
|
category
|
209126 |
disease_
|
Ontology term identifier for the disease, enabling standardized disease classification and referencing. Must be a term from the Mondo Disease Ontology (MONDO:) ontology term, or PATO:0000461 from the Phenotype And Trait Ontology (PATO:).
|
vector
|
category
|
209126 |
donor_
|
Identifier for the donor from whom the cell sample is obtained. |
vector
|
category
|
209126 |
is_
|
Indicates whether the data is primary (directly obtained from experiments) or has been computationally derived from other primary data. |
vector
|
bool
|
209126 |
self_
|
Ethnicity of the donor as self-reported, relevant for studies considering genetic diversity and population-specific traits. |
vector
|
category
|
209126 |
self_
|
Ontology term identifier for the self-reported ethnicity, providing a standardized reference for ethnic classifications. If the organism is human (organism_ontology_term_id == 'NCBITaxon:9606'), then the Human Ancestry Ontology (HANCESTRO:) is used.
|
vector
|
category
|
209126 |
sex
|
Biological sex of the donor or source organism, crucial for studies involving sex-specific traits or conditions. |
vector
|
category
|
209126 |
sex_
|
Ontology term identifier for the biological sex, ensuring standardized classification of sex. Only PATO:0000383, PATO:0000384 and PATO:0001340 are allowed.
|
vector
|
category
|
209126 |
size_
|
The size factors created by the normalisation method, if any. |
vector
|
float32
|
209126 |
soma_
|
If the dataset was retrieved from CELLxGENE census, this is a unique identifier for the cell. |
vector
|
int64
|
209126 |
suspension_
|
Type of suspension or medium in which the cells were stored or processed, important for understanding cell handling and conditions. |
vector
|
category
|
209126 |
tissue
|
Specific tissue from which the cells were derived, key for context and specificity in cell studies. |
vector
|
category
|
209126 |
tissue_
|
General category or classification of the tissue, useful for broader grouping and comparison of cell data. |
vector
|
category
|
209126 |
tissue_
|
Ontology term identifier for the general tissue category, aiding in standardizing and grouping tissue types. For organoid or tissue samples, the Uber-anatomy ontology (UBERON:) is used. The term ids must be a child term of UBERON:0001062 (anatomical entity). For cell cultures, the Cell Ontology (CL:) is used. The term ids cannot be CL:0000255, CL:0000257 or CL:0000548.
|
vector
|
category
|
209126 |
tissue_
|
Ontology term identifier for the tissue, providing a standardized reference for the tissue type. For organoid or tissue samples, the Uber-anatomy ontology (UBERON:) is used. The term ids must be a child term of UBERON:0001062 (anatomical entity). For cell cultures, the Cell Ontology (CL:) is used. The term ids cannot be CL:0000255, CL:0000257 or CL:0000548.
|
vector
|
category
|
209126 |
| var | ||||
feature_
|
Unique identifier for the feature, usually a ENSEMBL gene id. |
vector
|
object
|
28094 |
feature_
|
A human-readable name for the feature, usually a gene symbol. |
vector
|
object
|
28094 |
hvg
|
Whether or not the feature is considered to be a ‘highly variable gene’ |
vector
|
bool
|
28094 |
hvg_
|
A ranking of the features by hvg. |
vector
|
float64
|
28094 |
soma_
|
If the dataset was retrieved from CELLxGENE census, this is a unique identifier for the feature. |
vector
|
int64
|
28094 |
| obsp | ||||
knn_
|
K nearest neighbors connectivities matrix. |
sparsematrix
|
float32
|
209126 × 209126 |
knn_
|
K nearest neighbors distance matrix. |
sparsematrix
|
float64
|
209126 × 209126 |
| obsm | ||||
X_
|
The resulting PCA embedding. |
densematrix
|
float32
|
209126 × 50 |
| varm | ||||
pca_
|
The PCA loadings matrix. |
densematrix
|
float32
|
28094 × 50 |
| layers | ||||
counts
|
Raw counts |
sparsematrix
|
float32
|
209126 × 28094 |
normalized
|
Normalised expression values |
sparsematrix
|
float32
|
209126 × 28094 |
| uns | ||||
dataset_
|
Long description of the dataset. |
atomic
|
str
|
1 |
dataset_
|
A unique identifier for the dataset. This is different from the obs.dataset_id field, which is the identifier for the dataset from which the cell data is derived.
|
atomic
|
str
|
1 |
dataset_
|
A human-readable name for the dataset. |
atomic
|
str
|
1 |
dataset_
|
The organism of the sample in the dataset. |
atomic
|
str
|
1 |
dataset_
|
Bibtex reference of the paper in which the dataset was published. |
atomic
|
str
|
1 |
dataset_
|
Short description of the dataset. |
atomic
|
str
|
1 |
dataset_
|
Link to the original source of the dataset. |
atomic
|
str
|
1 |
knn
|
Supplementary K nearest neighbors data. |
dict
|
3 | |
normalization_
|
Which normalization was used |
atomic
|
str
|
1 |
pca_
|
The PCA variance objects. |
dict
|
2 | |