About multimodal single-cell data
The goal for this page is to provide an introduction to multimodal single-cell data and the specific datasets used in the competition. We know that many participants may be encountering this data type for the first time (it’s only been around for a couple of years!), and so this page serves as a general introduction. At the end you will find further reading recommendations that you can consult if you’d like to learn more.
The flow of genetic information in cells
Cells are the fundamental unit of life. All living things are made of of cells. This includes trees, fish, humans, bacteria, fungi, etc. Cells come in all shapes and sizes, but they have several properties in common no matter where we look. For example, all cells are membrane-bound. This means they have a clear boundary between inside and outside. All cells contain some form of organelles, which are specialized substructures that perform a specific function. And, all cells contain three layers of genetic information: DNA, RNA, and protein.

DNA defines an organism. Indeed, you can change the species of a bacteria through genome transplantation. However, DNA is not functional. It contains a set of instructions that must be converted into RNA and then into protein. For most purposes, you can think of RNA as a messenger between DNA and protein. DNA is made up of genes that contain instructions on how to make proteins. Proteins are responsible for carrying out biological functions in the cell, such as metabolising glucose to create energy for the cell. Generally speaking, each protein in your body is encoded by a single gene.
Although all of cells in your body contain the same genome, the same set of DNA, these trillions carry out very different biological functions. The differences between an immune cell, a neuron, or a muscle cell is defined by which genes are turned on or off within those cells. When a gene is turned on, more copies of RNA are created, thereby increasing the production of protein. We know that regulation of the amount of protein both happens at the level of transcription (DNA -> RNA) and translation (RNA -> protein).
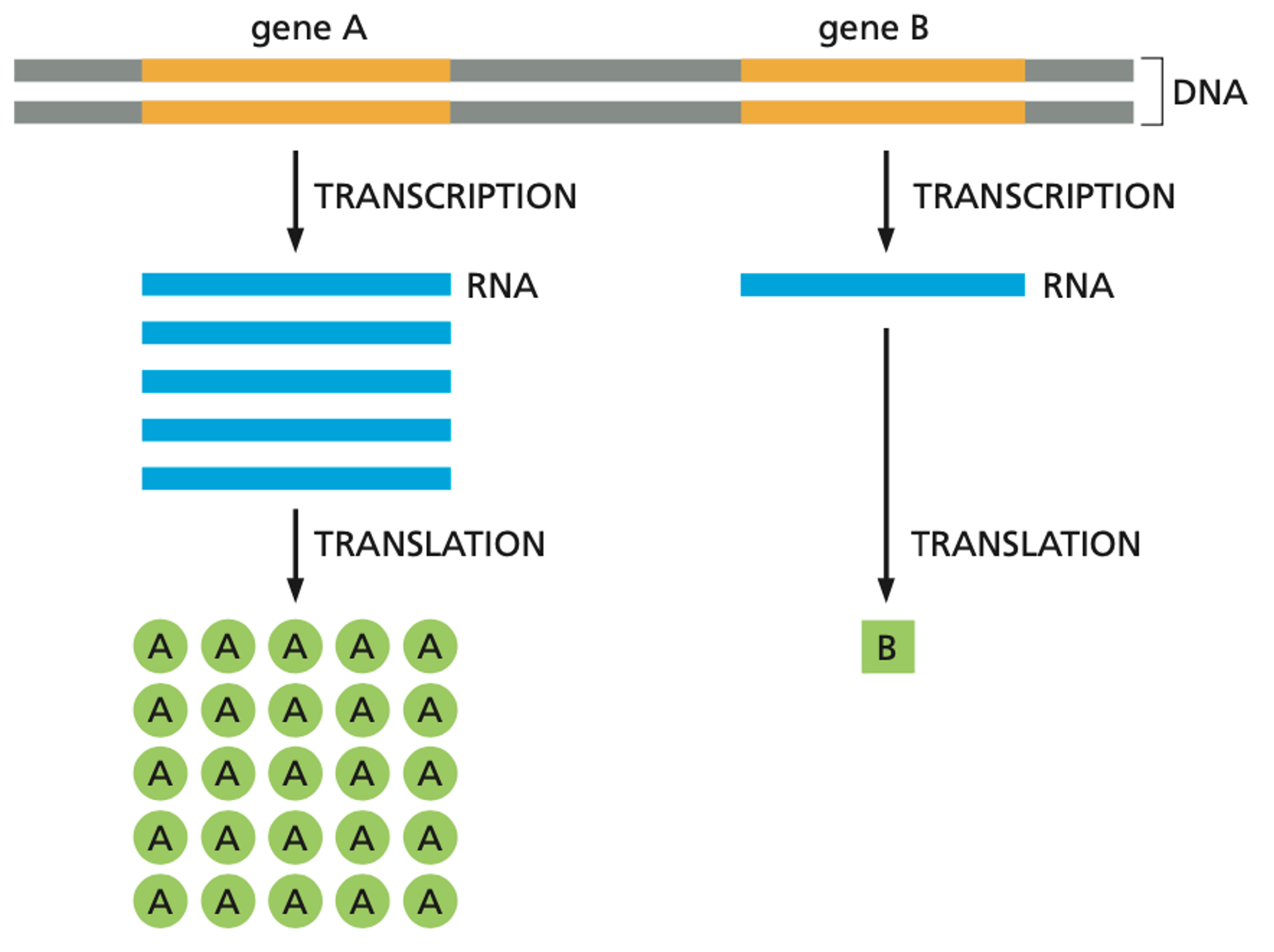
Because we know that the difference between types of cells has to do with different levels of RNA and proteins, it’s very useful to be able to measure the abundance of these molecules at the level of individual cells. Not only does this give us a fine-resolution view into the different kinds of cells in the body, it also provides insight into how the same set of DNA instructions can be interpreted so differently throughout the body.
The promise of single-cell measurements of genetics information is that by better understanding how this information flow within our cells and tissues, we might better understand what goes wrong in the context of disease.
A rough history of single-cell technologies
This is an exciting time to study single-cell data.
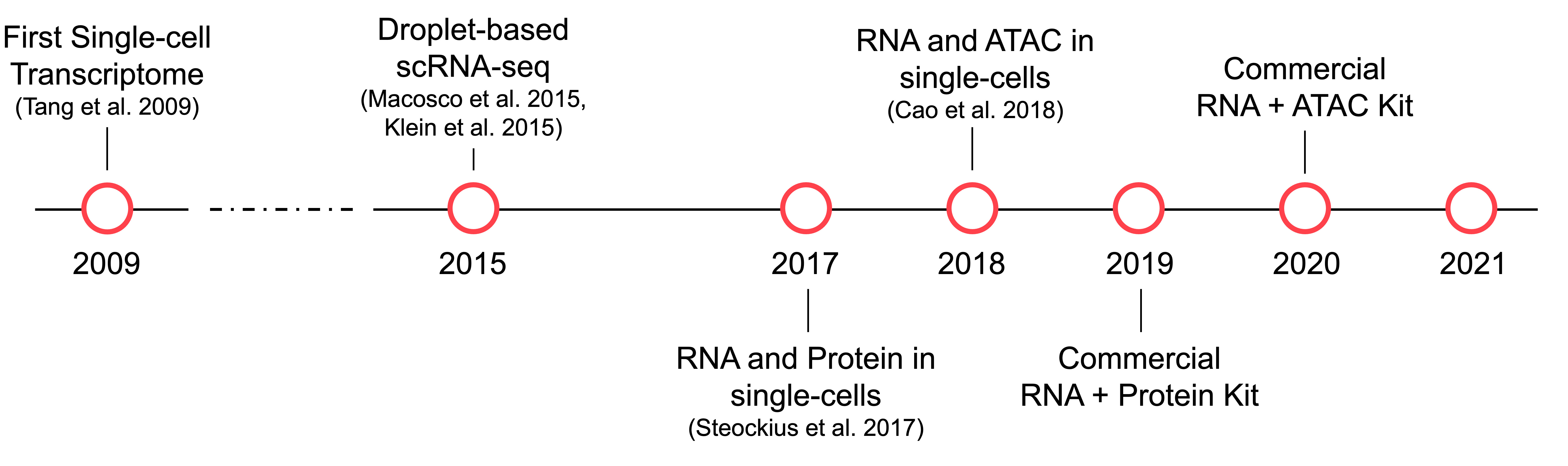
The first measurement of RNA from single cells was described in Eberwine et al. (1992) using molecular probes. It wasn’t until 2009, when Tang et al. described the sequencing of the transcriptome of a single cell (a mouse blastomere). In the following 6 years, several innovations were developed to improve the throughput of single-cell RNA sequencing (scRNA-seq). Perhaps the most impactful was the simultaneous description of two droplet-based protocols for capturing single cells into nanoliter droplets in oil emulsion described by Klein et al. (2015) and Macosko et al. (2015). With droplet-based single-cell methods, it became possible to perform experiments with tens of thousands of cells.

The next major innovation in single-cell measurement came in Steockius et al. (2017) and Cao et al. (2018) with the introduction of multimodal single-cell data measuring both RNA and protein or chromatin accessibility and RNA, respectively. The first method, called Cellular Indexing of Transcriptomes and Epitopes by Sequencing (CITE-seq), measures all RNA and expression of ~10-200 cell surface markers in individual cells. The second method, referred to as Multiome Assay for Transposase-Accessible Chromatin using sequencing (ATAC) + Gene Expression, provides a measure of chromatin accessibility throughout the genome and levels of gene expression for all genes. Unlike CITE-seq, this Multiome technology provides a view across all DNA and RNA in the cell. The following year, Nature Methods named Single-cell multimodal omics the Method of the Year 2019 noting these methods “opened unprecedented opportunity for the development of advanced statistical and computational methods.”
We are excited that in 2021, there are robust commercially-available reagents that we can use to create a reference benchmarking dataset to drive innovation in multimodal single-cell data integration.
Measuring multiple modalities in single cells
We know that DNA must be accessible (ATAC data) to produce mRNA (expression data), and mRNA in turn is used as a template to produce protein (protein abundance). These processes are regulated often by the same molecules that they produce: for example, a protein may bind DNA to prevent the production of more mRNA. Understanding these regulatory processes would be transformative for synthetic biology and drug target discovery. Any method that can predict a modality from another must have accounted for these regulatory processes, but the demand for multi-modal data shows that this is not trivial.

It is also powerful to be able to capture both RNA and protein expression in the same cell. Proteins on the cell surface are not only important for identifying different cell populations, but these proteins also serve functional roles, especially within the immune system. Currently, it is only possible to measure roughly 100-200 proteins on an individual cell, but we know cells contain tens of thousands (or more) unique proteins. Nevertheless, this information has shown crucial for disentangling the identities of cell populations that are transcriptionally similar but express different functional surface markers.

Multimodal data as a basis for benchmarking
Developing machine learning methods for biological systems is complicated by the difficulty of obtaining ground truth. Typically, machine learning tasks rely on manual annotation (as in images or natural language queries), dynamic measurements (as in longitudinal health records or weather), or multimodal measurement (as in translation or text-to-speech). However, this is more complicated in the context of single-cell biology.
With single-cell data, annotation isn’t feasible. The data is noisy and not fully understood with descriptions of cell types evolving rapidly. Similarly, longitudinal measurement of all the RNA in a cell isn’t possible because the current measurement technologies involve destroying the cell. However, with multimodal single-cell data, we can now directly observe two layers of genetic information in the same cells. This provides an opportunity to use the fact these two sets of data were observed co-occurring in the same cells as ground truth. This is akin to the way that access to the same sentiment expressed in two languages provides ground truth for machine translation.
However, as these technologies are relatively new, most publicly available datasets are designed for exploration, not benchmarking. To set up a competition for multimodal single-cell data integration, we set out to create a fit-for-purpose benchmarking dataset.